Isochromosome
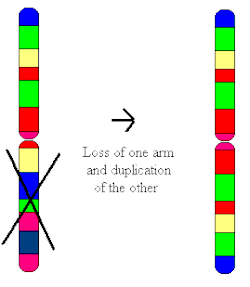
Un isochromosome est une anomalie structurelle déséquilibrée dans laquelle les bras du chromosome sont des images miroir l'un de l'autre[1]. Le chromosome est constitué de deux copies du bras long (q) ou du bras court (p), car la formation d'isochromosomes équivaut à une duplication et une suppression simultanées du matériel génétique. Par conséquent, il existe une trisomie partielle des gènes présents dans l’isochromosome et une monosomie partielle des gènes dans le bras perdu[2].
Nomenclature
Un isochromosome est une anomalie chromosomique qui peut être désignée de manière concise en utilisant une nomenclature spécifique. Cette abréviation se compose généralement de la lettre "i" suivie du numéro du chromosome impliqué, puis d'une indication du point de rupture centromérique entre parenthèses. Par exemple, si nous avons un isochromosome qui dérive du chromosome 17 et qu'il est constitué de deux bras longs (bras "q"), il sera alors identifié de façon précise comme i(17)(q10). Cette notation permet de communiquer de manière standardisée la nature de cette réorganisation chromosomique)[3].
Mécanisme
Les isochromosomes peuvent être créés au cours de la mitose et de la méiose par une mauvaise division du centromère ou un échange de brins de type U.[1]
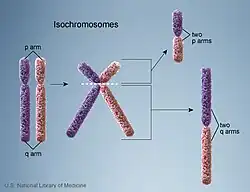
Mauvaise division du centromère
Lors d'une séparation normale des chromatides sœurs en anaphase, le centromère se divise longitudinalement, ou parallèlement à l'axe long du chromosome[4]. Un isochromosome est créé lorsque le centromère est divisé transversalement, ou perpendiculairement à l'axe long du chromosome. La division ne se produit généralement pas dans le centromère lui-même, mais dans une zone entourant le centromère, également appelée région péricentrique[2]. Il est proposé que ces sites d’échange contiennent des séquences homologues entre chromatides sœurs[5] Bien que le chromosome résultant puisse paraître monocentrique avec un seul centromère, il est isodicentrique avec deux centromères très proches l'un de l'autre ; ce qui entraîne une perte potentielle de matériel génétique présent sur les autres bras[2],[5]. Une mauvaise division du centromère peut également produire des isochromosomes monocentriques, mais ils ne sont pas aussi courants que les isochromosomes dicentriques[1],[6].
Échange de brins de type U
Un mécanisme plus courant dans la formation des isochromosomes est la rupture et la fusion des chromatides sœurs, se produisant très probablement au début de l'anaphase de la mitose ou de la méiose[4]. Une cassure double brin dans la région péricentrique du chromosome est réparée lorsque les chromatides sœurs, chacune contenant un centromère, sont fusionnées[2]. Une mauvaise division du centromère et un échange de type U peuvent se produire dans les chromatides sœurs, créant ainsi un isochromosome avec des bras génétiquement identiques. Cependant, l'échange de type U peut également se produire pour les chromosomes homologues, ce qui crée un isochromosome avec des bras homologues. Cet échange entre homologues est très probablement dû à des séquences homologues contenant un faible nombre de répétitions . Quel que soit le chromosome impliqué dans l'échange de type U, le fragment acentrique du chromosome est perdu, créant ainsi une monosomie partielle de gènes situés dans cette partie du chromosome acentrique[2].
Conséquences
L'isochromosome le plus courant est le chromosome sexuel X.[5] Les chromosomes autosomiques acrocentriques 13, 14, 15, 21 et 22 sont également des candidats courants pour la formation d'isochromosomes[1]. Les chromosomes contenant des bras plus petits sont plus susceptibles de devenir des isochromosomes car la perte de matériel génétique dans ces bras peut être tolérée[2].
syndrome de Turner
Le syndrome de Turner est une maladie qui touche les femmes et qui se caractérise par la perte partielle ou totale d’un chromosome X. Cela provoque des symptômes tels que des problèmes de croissance et de développement sexuel. Chez 15 % des patients atteints du syndrome de Turner, l'anomalie structurelle est l'isochromosome X, qui est composé de deux copies du bras q (i(Xq))[1],[2]. La majorité des i(Xq) sont créés par échange de brins de type U. Une rupture et une réunion dans la région péricentrique du bras p donnent lieu à un isochromosome dicentrique[5]. Une partie du bras p peut être trouvée dans cette formation de i(Xq), mais la majorité du matériel génétique du bras p est perdue, il est donc considéré comme absent. Étant donné que le bras p du chromosome X contient des gènes nécessaires au développement sexuel normal, les patients atteints du syndrome de Turner subissent des effets phénotypiques qui s'en retrouves déformées[4]. Alternativement l'augmentation du dosage des gènes sur le bras q peut être impliquée dans une augmentation de 10 fois du risque que les patients atteints de la maladie de Turner i(Xq) développent une thyroïdite auto-immune, une maladie dans laquelle le corps crée des anticorps pour cibler et détruire les cellules thyroïdiennes[7].
Néoplasie
La néoplasie est une croissance cellulaire incontrôlée, entraînant la création d’une tumeur. Dans de nombreuses formes différentes de néoplasie, l'isochromosome 17q est l'isochromosome le plus fréquemment associé à la néoplasie et correspond à une faible survie des patients[8],[9]. Des séquences d'ADN uniques, appelées répétitions à faible nombre de copies, se produisent dans la région péricentrique du bras p, de sorte qu'un événement de croisement dans cette zone peut créer un isochromosome dicentrique par échange de brins de type U.[10] La néoplasie créée à partir de i(17q) est causée par une diminution et une augmentation du dosage génétique de la monosomie du bras p et de la trisomie du bras q . De nombreux gènes suppresseurs de tumeurs candidats sont trouvés sur le bras p perdu, permettant de maintenir la population de cellules tumorales[9]. On se demande si la perte du gène suppresseur de tumeur p53, situé sur 17p, est impliquée dans la pathogenèse centrale de certaines néoplasies. La présence d'un gène p53 peut être fonctionnellement active, mais sa relation avec d'autres oncogènes peut modifier ses niveaux d'expression lorsqu'il n'est présent qu'en une seule copie[8],[9],[10]. Étant donné l'étendue significative des séquences génétiques qui sont associées à la néoplasie i(17q), il est particulièrement complexe d'identifier avec précision les gènes spécifiques, ou même la combinaison exacte de gènes, qui contribuent activement à la croissance et au développement de la tumeur. En effet, la taille de ces régions rend difficile le ciblage des éléments génétiques clé responsables du processus tumoral.
Références
- 1 2 3 4 5 editors, Roger N. Rosenberg ... [et al., The molecular and genetic basis of neurologic and psychiatric disease, Philadelphia, 4th, (ISBN 978-0781769563), p. 22
- 1 2 3 4 5 6 7 Steven L. Gersen, Martha B. Keagle editors, The principles of clinical cytogenetics, New York, 3rd, (ISBN 978-1441916884)
- ↑ « Dictionnaire médical de l'Académie de Médecine », sur www.academie-medecine.fr (consulté le )
- 1 2 3 Manu L. Kothari, Lopa A. Mehta, Sadhana S. Roychoudhury, Essentials of human genetics, Hyderabad, India, 5th, (ISBN 978-8173716478)
- 1 2 3 4 Wolff, Miller, Van Dyke et Schwartz, « Molecular definition of breakpoints associated with human Xq isochromosomes: implications for mechanisms of formation », Am J Hum Genet, vol. 58, no 1, , p. 154–160 (PMID 8554051, PMCID 1914957)
- ↑ « Cours », sur archives.uness.fr (consulté le )
- ↑ Elsheikh, Wass et Conway, « Autoimmune thyroid syndrome in women with Turner's syndrome-the association with karyotype », Clinical Endocrinology, vol. 55, no 2, , p. 223–226 (PMID 11531929, DOI 10.1046/j.1365-2265.2001.01296.x, S2CID 73303081)
- 1 2 Cancer cytogenetics: chromosomal and molecular genetic aberration of tumour cells, 4, , 22, 94 (ISBN 978-1118795514)
- 1 2 3 Mendrzyk, Korshunov, Toedt et Schwarz, « Isochromosome breakpoints on 17p in medulloblastoma are flanked by different classes of DNA sequence repeats », Genes, Chromosomes and Cancer, vol. 45, no 4, , p. 401–410 (PMID 16419060, DOI 10.1002/gcc.20304, S2CID 20113481)
- 1 2 Barbouti, Stankiewicz, Nusbaum et Cuomo, « The Breakpoint Region of the Most Common Isochromosome, i(17q), in Human Neoplasia Is Characterized by a Complex Genomic Architecture with Large, Palindromic, Low-Copy Repeats », American Journal of Human Genetics, vol. 74, no 1, , p. 1–10 (PMID 14666446, PMCID 1181896, DOI 10.1086/380648)
 Portail de la biologie
Portail de la biologie  Portail de la médecine
Portail de la médecine  Portail de la biologie cellulaire et moléculaire
Portail de la biologie cellulaire et moléculaire